

Last Updated: October 31, 2025
Introduction to Lesch-Nyhan Syndrome
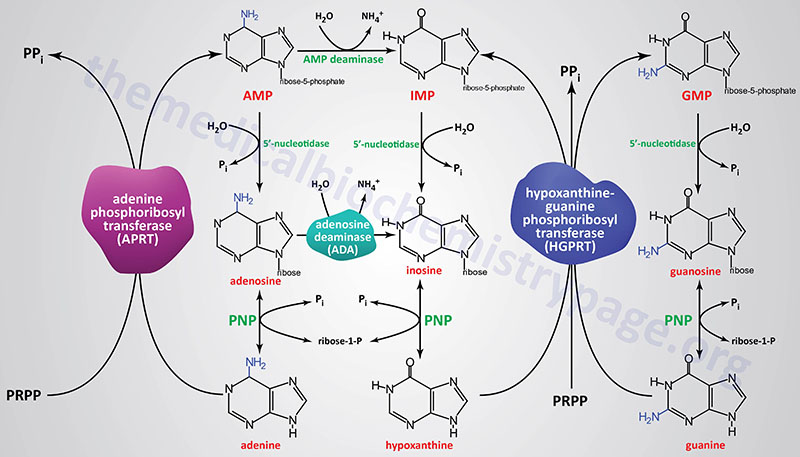
Lesch-Nyhan syndrome (LNS) is an X-linked recessive disorder resulting from the near complete lack of activity of the purine nucleotide salvage enzyme, hypoxanthine-guanine phosphoribosyltransferase, HGPRT (encoded by the HPRT1 gene). The HGPRT enzyme catalyzes the following two interconversions:
hypoxanthine + PRPP ↔ IMP + PPi
guanine + PRPP ↔ GMP + PPi
There are three over-lapping clinical syndromes associated with deficiencies in HGPRT activity. Individuals that have less than 1.5% residual enzyme activity exhibit debilitating neurologic disability, behavioral abnormalities that include impulsive and self-mutilating behaviors and varying degrees of cognitive disability in addition to overproduction of uric acid. This most severe of the three clinical syndromes is Lesch-Nyhan syndrome.
The second clinical syndrome represents patients who have 1.5% to 8% of residual enzyme activity. In this second grouping, patients exhibit neurologic disability that ranges from clumsiness to debilitating pyramidal (CNS neurons involved in voluntary motor movement) and extrapyramidal motor dysfunction in addition to overproduction of uric acid.
In the third clinical grouping patients have at least 8% of normal HGPRT activity. This last grouping of patients exhibit overproduction of uric acid and associated hyperuricemia, renal lithiasis (kidney stones) and gout. The latter circumstance (partial deficiency with at least 8% enzyme activity) is associated with Kelley-Steegmiller syndrome.
Molecular Biology of Lesch-Nyhan Syndrome
Lesch-Nyhan syndrome is inherited as an X-linked recessive disorder with an incidence of approximately 1:380,000. Since it is an X-linked disease it is found almost exclusively in males although affected (manifesting) females have been identified, albeit very rarely. The HPRT1 gene is located on the X chromosome (Xq26.2–q26.3) spanning 44 kbp and composed of 9 exons that encode a protein of 218 amino acids.
Over 270 different mutations in the HPRT1 gene have been identified in Lesch-Nyhan syndrome patients. Alterations to the gene include single base insertions and deletions, large deletions, amino acid substitutions (missense mutations), and stop codon mutations (nonsense mutations). Single point mutations are the main cause of partial deficiency of the enzyme, whereas Lesch-Nyhan syndrome is predominantly the result of mutations that modify the size of the predicted protein. About 30% of Lesch-Nyhan patients’ mothers are not somatic carriers, and these patients probably carry de novo mutations due to a germinal cell mutation.
Clinical Features of Lesch-Nyhan Syndrome
The characteristic clinical features of the Lesch-Nyhan syndrome are intellectual impairment, spastic cerebral palsy, choreoathetosis, uric acid urinary stones, and self-destructive biting of fingers and lips. The overall clinical features of LNS can be divided into three broad categories. These include uric acid overproduction and its associated consequences (e.g. gouty arthritis and renal lithiasis), neurobehavioral dysfunction indicative of central nervous system involvement, and growth impairment. As indicated (and as expected from uric acid overproduction) LNS patients manifest with many of the symptoms of classic gout and will not be covered here.
All patients with Lesch-Nyhan syndrome manifest with profound motor dysfunction that is recognizable within the first 3 to 9 months of life. Infants fail to develop the ability to hold up their heads or to sit unaided. Further motor development will be delayed and the onset of pyramidal and extrapyramidal signs become evident by 1 to 2 years of age. In LNS patients there are three major signs of pyramidal dysfunction: spasticity, hyperreflexia and the extensor plantar reflex (also known as the Babinski reflex: the great toe flexes toward the top of the foot and the other toes fan out after the sole of the foot has been firmly stroked). Extrapyramidal dysfunction in LNS patients is primarily evident as dystonia (sustained muscle contractions causing twisting and repetitive movements or abnormal postures) although many patients also exhibit choreoathetosis (involuntary movement disorder in association with slow continuous writhing particularly of the hands and feet).
Most commonly associated with Lesch-Nyhan syndrome is the behavioral dysfunction manifest with impulsivity and self-mutilation particularly of the lips, fingers and tongue. LNS patients will often strike out at people around them, spit on people and use foul language. These latter symptoms are analogous to the uncontrollable compulsions associated with Tourette syndrome. The self-injury behavior is clearly an involuntary action as most LNS patients will learn to call out for help when they feel the compulsive behavior overtaking them, or they will sit on their hands or wear socks or gloves to limit the self-injurious behavior. Although the precise cause of the self-mutilating behavior in LNS patients is not clearly understood it is most likely that it is a form of obsessive-compulsive disorder. In many circumstance the routine surgical removal of a LNS patient’s teeth is considered most beneficial.