
Last Updated: November 9, 2025
Introduction to Gaucher Disease
Gaucher disease (pronounced “go-shay”) belongs to the family of disorders identified as lysosomal storage disorders. The disease is named for the French physician, Philippe Gaucher, who first described the disorder in 1882. Gaucher disease is an autosomal recessive disorder that is characterized by the lysosomal accumulation of glucosylceramide (glucocerebroside) which is a normal intermediate in the catabolism of globosides and gangliosides. Due to the high levels of glucocerebrosides in the lysosomes of cells of the monocyte/macrophage lineage these cells are the major contributors to the pathology of Gaucher disease.
Molecular Biology of Gaucher Disease
Gaucher disease results from defects in the gene encoding the lysosomal hydrolase, glucosylceramidase beta 1, also called acid β-glucosidase or more commonly glucocerebrosidase.
Humans express three glucosylceramidase genes identified as GBA1, GBA2, and GBA3. The enzyme encoded by the GBA2 gene is localized to the microsomes where it catalyzes the hydrolysis of bile acid 3-O-glucosides. The enzyme encoded by the GBA3 gene is localized to the cytosol where it hydrolyzes several different glycosides.
Glucosylceramidase beta 1 is encoded by the GBA1 gene. The GBA1 gene is located on chromosome 1q22 and spans 7 kb encompassing 12 exons that generate five alternatively spliced mRNAs. These five mRNAs encode three distinct isoforms of glucocerebrosidase. Isoform 1 is 536 amino acids, isoform 2 is 449 amino acids, and isoform 3 is 487 amino acids.
Glucosylceramidase beta 1 exists as a homodimer in human cells. In addition to the enzyme itself, an active hydrolytic complex requires an additional activator protein. The activator of glucosylceramidase beta 1 is saposin C, a member of the saposin family of small glycoproteins.
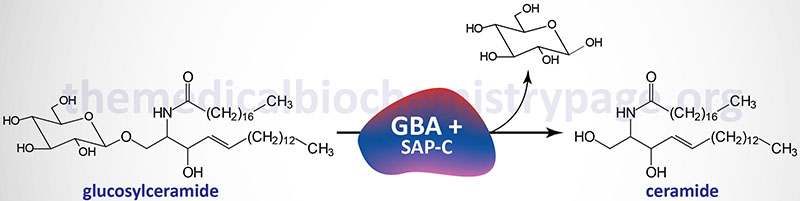
The natural substrates for glucosylceramidase beta 1 are N-acyl-sphingosyl-1-O-β-D-glucosides, glucosylceramides, and various sphingosyl compounds. The physiological significance of the role of saposin C in glucosylceramidase beta 1 activity was demonstrated in patients with a saposin C deficiency exhibiting a Gaucher-like disease phenotype.
As of 2025 there have been a total of 106 pathogenic mutations identified in the GBA1 gene that are associated with the development of Gaucher disease. The majority of mutations leading to Gaucher disease are missense mutations. A range of other types of mutation have also been described in Gaucher disease including frameshift mutations, splicing mutations, deletions, and gene fusions. The most commonly occurring mutation in the US is found in the Ashkenazi Jewish population at nucleotide 1226 resulting in the substitution of asparagine (N) at position 370 for serine (S). This mutation is identified as the N370S mutation. This mutation is associated with type 1 Gaucher disease only. Another mutation at nucleotide 1448, resulting in a leucine to proline change at amino acid 444 (L444P), is associated with the neuronopathic forms of Gaucher disease.
Prosaposin
The saposins (A, B, C, and D) are all derived from a single precursor, prosaposin. Prosaposin is encoded by the PSAP gene. The PSAP gene is located on chromosome 10q22.1 and is composed of 15 exons that generate three alternatively spliced mRNAs, each of which encoded a distinct preproprotein isoform. The isoform a preproprotein is composed of 524 amino acids. The isoform b preproprotein is composed of 527 amino acids. The isoform c preproprotein is composed of 526 amino acids.
The mature saposins, as well as prosaposin, activate several lysosomal hydrolases involved in the metabolism of various sphingolipids. Prosaposin is proteolytically processed to saposins A, B, C and D, within lysosomes but also exists as an integral membrane protein not destined for lysosomal entry. Native, unprocessed prosaposin can be found in many biological fluids such as seminal plasma, human milk, and cerebrospinal fluid, where it appears to have a different function.
Saposin A, along with saposin C, is a cofactor for the function of the GALC encoded enzyme, galactosylceramidase (also called galactocerebrosidase). Mutations in the PSAP gene that affect the production of saposin A and saposin C are associated with the development Krabbe disease which is primarily caused by mutations in the GALC gene.
Saposin B is a cofactor for the function of the GLB1 encoded enzyme, β-galactosidase-1. Mutations in the PSAP gene that affect the production of saposin B are associated with the development of Morquio Syndrome type B, (mucopolysaccharidosis IVB; MPS IVB) which is primarily the result of mutations in the GLB1 gene.
Saposin C, as described above, is a cofactor for the function of the GBA1 encoded glucocerebrosidase enzyme and as such, mutations in the PSAP gene that affect the production of saposin C are associated with the development of Gaucher disease.
Saposin D is a cofactor for the ASAH1 (N-acylsphingosine amidohydrolase 1) encoded enzyme commonly called acid ceramidase. Mutations in the PSAP gene that affect the production of saposin D are associated with the development of Farber lipogranulomatosis, a lysosomal storage disorder (lysosomal storage disease) primarily caused by mutations in the ASAH1 gene.
Clinical Features of Gaucher Disease
The hallmark of Gaucher disease is the accumulation of lipid-engorged cells of the monocyte/macrophage lineage with a characteristic appearance in a variety of tissues. The primarily affected tissues are bone, bone marrow, spleen, and liver. These distinctive cells contain one or more small nuclei and their cytoplasm contains a coarse fibrillary material that forms a striated tubular pattern often described as “wrinkled tissue paper”. These cells are called Gaucher cells. Gaucher cells also induce the production of pro-inflammatory cytokines resulting in enlargement of the liver and spleen (hepatosplenomegaly), destruction of bone, lung abnormalities, anemia, leukopenia, and thrombocytopenia.
Clinically, Gaucher disease is classified into three major types. These types are determined by the absence or presence and severity of neurological involvement. Type 1 is the most commonly occurring form of Gaucher disease and is called the non-neuronopathic type (historically called the adult form). Type 1 Gaucher disease represents 95% of all cases of the disease. Both type 2 and type 3 Gaucher disease have neuronopathic involvement. Type 2 disease is the acute neuronopathic form, exhibits early onset of severe central nervous system dysfunction and is usually fatal within the first 2 years of life. Type 3 Gaucher disease (subacute neuronopathic) patients have later onset neurological symptoms with a more chronic course than type 2 patients.
As indicated , enlargement of the liver is characteristic in all Gaucher disease patients. In severe cases the liver can fill the entire abdomen. Splenomegaly is present in all but the most mildly affected individuals and even in asymptomatic individuals spleen enlargement can be found. In addition to hepatosplenomegaly, bleeding is a common presenting symptom in Gaucher disease. The most common cause of the bleeding is thrombocytopenia (deficient production of platelets).
Type 1 (adult, nonneuronopathic)
Type 1 Gaucher disease has a broad spectrum of severity from early onset of massive hepatosplenomegaly and extensive skeletal abnormalities to patients lacking symptoms until the eighth or ninth decade of life. The median age of appearance of symptoms in type 1 Gaucher disease is 30 years. Almost all mildly affected type 1 patients harbor the N370S mutation. The symptoms of type 1 Gaucher disease result from engorged macrophages that result in hepatosplenomegaly with resultant dysfunction of the liver and spleen.
Type 2 (infantile, neuronopathic)
Type 2 Gaucher disease is characterized by onset at an early age (hence being called the infantile form) and severe neurologic involvement. Extensive visceral involvement with hepatosplenomegaly is characteristic of type 2 Gaucher disease. Abnormalities in oculomotor function is often the first manifesting symptom in type 2 disease. Patients often thrust their heads in an attempt to compensate when following a moving object. An ichthyosis-like (dry scaly skin) disorder has been suggested as a symptom to allow for the differentiation of type 2 Gaucher disease from types 1 and 3.
Type 3 (juvenile, neuronopathic)
Type 3 Gaucher disease is divided into three sub-classes. Type 3a presents with progressive neurologic involvement dominated by dementia and myoclonus (involuntary muscle twitching). Type 3b presents with aggressive skeletal and visceral symptoms. The neurological symptoms are limited to horizontal supranuclear gaze palsy. Type 3c presents with neurological involvement limited to horizontal supranuclear gaze palsy, cardiac valve calcification and corneal opacities but with little visceral involvement.
Treatment of Gaucher Disease
Currently there is no effective treatment for type 2 Gaucher disease. Current treatments for type 1 and type 3 Gaucher disease include enzyme replacement therapy (ERT), bone marrow transplantation (BMT), or oral medications. ERT replaces the deficient enzyme with artificial enzymes. The biotech company Genzyme, a unit of Sanofi SA, markets the drug imiglucerase (Cerezyme) for ERT treatment of Gaucher disease.
Two additional ERT drugs prescribed for type 1 Gaucher patients are velaglucerase alpha (VPRIV) and taliglucerase alpha (Elelyso). These replacement enzymes are administered in an outpatient procedure through a vein (intravenously), typically in high doses at two-week intervals. Although results can vary, treatment is frequently effective in people with type 1 Gaucher disease and, in some cases, type 3.
In many people, enzyme replacement therapy can reduce the enlargement of the liver and spleen, help to resolve blood abnormalities and improve bone density. It’s unclear whether this therapy is effective for the neurological problems of Gaucher disease. Occasionally people experience an allergic or hypersensitivity reaction to enzyme treatment.
BMT has been used for severe cases of Gaucher disease. In this technique, blood-forming cells that have been damaged by Gaucher are removed and replaced, which can reverse many of Gaucher signs and symptoms. Because this is a high-risk approach, it’s performed less often than is enzyme replacement therapy.
The oral medication miglustat (Zavesca) has been approved for use in people with Gaucher disease. It appears to interfere with the production of glucocerebrosides in some people with type 1 disease. Diarrhea and weight loss are common side effects. This medication may also affect sperm production. Contraception is advised while using miglustat and for three months after stopping the drug.